Formats#
Note
Un grand merci à Matthias Zytnicki !
FASTA#
Type de fichier#
Séquence
Signification du nom#
Format utilisé par l’outil FastA (fast alignment)
Qui le génère#
Presque tous
Qui le lit#
Presque tous, vous
Exemple#
>sequence1
CGATGTACGCTAGAT
Explications#
Chaque séquence commence par un chevron (>), suivi du nom de la séquence. Bien que cela ne soit pas obligatoire, il est recommandé que le nom de la séquence soit unique dans le fichier. La séquence elle-même suit.
FASTQ#
Type de fichier#
Séquence de lecture
Signification du nom#
Comme FASTA, mais avec la qualité (Q)
Qui le génère#
Le séquenceur
Qui le lit#
Les outils de mapping, les visualisateurs, vous
Exemple#
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Explications#
Chaque séquence est codée sur 4 ligne : un @, suivi du nom de la séquence ; la séquence elle-même suit ; un + (avec éventuellement le nom de la séquence, encore une fois) ; la qualité de la séquence. La qualité de la séquence suit un codage particulier, où chaque caractère représente un nombre. En général, l’association est la suivante:
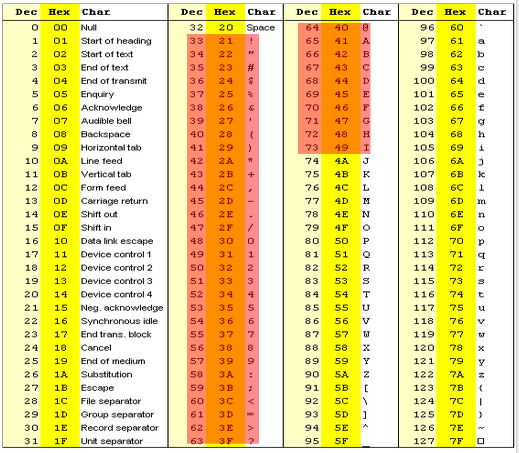
!correspond à 0“correspond à 1#correspond à 2$correspond à 3…
Icorrespond à 40
Chaque nombre représente la probabilité p de se tromper sur la lecture d’une base. Le code représente la valeur −10 log10(p). Par exemple, le caractère C code le nombre 34. Il représente donc une probabilité d’erreur d’environ 4.10-4. Les codes les plus à droite représentent donc les meilleures qualités. Attention : pour des données relativement anciennes, il existe d’autres codages de la qualité (i.e. d’autres associations entre les caractères et les nombres).
See also
Pour en savoir plus
Page Wikipedia du format : http://en.wikipedia.org/wiki/FASTQ_format
Référence : http://maq.sourceforge.net/fastq.shtml
Article NAR présentant le format : http://nar.oxfordjournals.org/content/38/6/1767.full
BED#
Type de fichier#
Annotation
Signification du nom#
Browser Extensible Format
Qui le génère#
Les outils d’annotation, TopHat
Qui le lit#
Les visualisateurs, vous
Remarque#
Le format BED est un format multi-forme, qui peut être utilisé dans beaucoup de contextes.
Exemple 1 (simple)#
chr1 100 200 peak_1 123
Explications#
Chaque ligne est une annotation. Les informations sont tabulées, i.e. chaque ligne contient un nombre fixe de colonnes (ici, 5), séparées par des tabulations.
Le format BED est utilisée pour beaucoup de types d’annotations, comme les régions MACS :
(chr1) le nombre du chromosome (ou du scaffold)
(100) position extrémale en 5’
(2099) position extrémale en 3’
(peak_1) nom systématique de la jonction
(123) score de la région
Dans d’autres contexte, on peut ne trouver que les 3 ou 4 premiers champs.
Exemple 2 (jonctions entre exons)#
chr1 990 2099 JUNC00001560 3 + 990 2099 255,0,0 2 40,67 0,1042
Explications#
Ici, le format contient 12 colonnes: 6. (chr1) le nombre du chromosome (ou du scaffold) 7. (990) position extrémale en 5’ des lectures chevauchant la jonction 8. (2099) position extrémale en 3’ des lectures chevauchant la jonction 9. (JUNC00001560) nom systématique de la jonction 10. (3) nombre de lectures couvrant la jonction 11. (+) brin 12. (990) même chose que la colonne 2 13. (2099) même chose que la colonne 3 14. (255,0,0) pas important 15. (2) pas important 16. (40,67) taille maximum des lectures couvrant l’exon à gauche et à droite de l’intron. 17. (0,1042) pas important

GTF#
Type de fichier#
Annotation
Signification du nom#
Gene Transfer Format
Qui le génère#
Les outils d’annotation
Qui le lit#
Les browsers, TopHat
Exemple#
chr20 example exon 100 200 . + . gene_id "g1"; transcript_id "t1";
chr20 example exon 300 400 . + . gene_id "g1"; transcript_id "t1";
chr20 example exon 500 600 . + . gene_id "g1"; transcript_id "t1";
chr20 example exon 100 450 . + . gene_id "g1"; transcript_id "t2";
Explications#
C’est un autre format tabulé à 9 champs. Chaque exon est noté sur une ligne :
(chr20) le chromosome
(example) la source de l’annotation, habituellement l’outil qui a généré l’annotation
(exon) le type d’annotation ; nous avons ici des exons, mais cela pourrait des CDS (si l’on s’intéressait aux protéines traduites)
(100) le début de l’annotation
(200) la fin de l’annotation
(.) pas important
(+) le brin
(.) pas important
(gene_id “g1”; transcript_id “t1”;) les attributs, ou champs. C’est un fourre-tout. On peut y trouver le nom usuel du gène.
Le champ 9 indique à quel transcrit et quel gène appartient chaque exon.

See also
Pour en savoir plus
Documentation : http://mblab.wustl.edu/GTF22.html
Le format GTF est adapté du format GFF, moins contraint. Documentation du format GFF : http://www.sequenceontology.org/gff3.shtml
GFF#
Type de fichier#
Annotation
Signification du nom#
Gene Feature Format
Qui le génère#
Les outils d’annotation
Qui le lit#
Les browsers
Exemple#
chr20 example exon 100 200 . + . gene_id "g1"; transcript_id "t1";
chr20 example exon 300 400 . + . gene_id "g1"; transcript_id "t1";
chr20 example exon 500 600 . + . gene_id "g1"; transcript_id "t1";
chr20 example exon 100 450 . + . gene_id "g1"; transcript_id "t2";
Explications#
C’est un autre format tabulé à 9 champs. Chaque exon est noté sur une ligne :
(chr20) le chromosome
(example) la source de l’annotation, habituellement l’outil qui a généré l’annotation
(exon) le type d’annotation ; nous avons ici des exons, mais cela pourrait des CDS (si l’on s’intéressait aux protéines traduites)
(100) le début de l’annotation
(200) la fin de l’annotation
(.) pas important
(+) le brin
(.) pas important
(gene_id “g1”; transcript_id “t1”;) les attributs, ou champs. C’est un fourre-tout. On peut y trouver le nom usuel du gène.
SAM#
Type de fichier#
Mapping
Signification du nom#
Sequence Alignment/Map
Qui le génère#
Les outils de mapping
Qui le lit#
Vous, samtools
Exemple#
@SQ SN:chr1 LN:45
r001 99 chr1 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG *
r002 0 chr1 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *
r003 0 chr1 9 30 5S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0;
r004 0 chr1 16 30 6M14N5M * 0 0 ATAGCTTCAGC *
r003 2064 chr1 29 17 6H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1;
r001 147 chr1 37 30 9M = 7 -39 CAGCGGCAT * NM:i:1
Explications#
Le format SAM se décompose en deux parties : l’en-tête et le corps.
L’en-tête donne des informations sur le génome ou le sur le mapping. Les lignes d’en-tête commencent toutes par un @, suivi de deux lettres. La ligne @SQ SN:chr1 LN:45 se lit :
@: nous sommes dans un en-têteSQ: qui a trait à une séquence de référence (les chromomes)SN:chr1: le nom d’une séquence est chr1LN:45: sa taille est de 45 pb
Il existe beaucoup de type d’en-tête différents, que nous ne verrons pas. Le corps est un format tabulé :
(r001) le nom d’une lecture
(99) informations binaires sur la lecture. Un unique chiffre enregistre les informations suivantes : fragment mappé ou non, fragment d’un paired-end (ou d’un single-end), premier de la paire, etc. Pour savoir ce que signifie un nombre, il suffit d’aller sur le site http://broadinstitute.github.io/picard/explain-flags.html.
(chr1) la séquence sur lequel est mappée la lecture; on utilise * si la lecture n’est pas mappée
(7) position la plus 5’ de la lecture 5.(30) qualité du mapping, soit −10 log10(p), où p est la probabilité, estimée par l’outil de mapping, que la lecture soit mappée à la mauvaise place
(8M2I4M1D3M) format CIGAR de la lecture (cf infra)
(=) séquence sur lequel est mappé l’autre fragment, en cas de paired-end ; on utilise = si le chromosome est le même, et * si la lecture est single-end
(37) en cas de paired-end, osition la plus 5’ de l’autre fragment ; on utilise 0 si l’on est en single-end, si l’autre fragment ne mappe pas, etc.
(39) en cas de paired-end, taille de la lecture, soit la différence entre la position la plus 5’ du fragment 5’ et la position la plus 3’ du fragment 3’
(TTAGATAAAGGATACTG) séquence du fragment
(*) qualité du fragment (similaire au FASTQ) ; on utilise * si on ne souhaite pas renseigner ce champ
(SA:Z:ref,29,-,6H5M,17,0;) autres informations (cf infra)
Voici l’alignement des lectures correspondant au fichier SAM.
Coor 11111 1111122222222223333333333444444
12345678901234 5678901234567890123456789012345
chr1 AGCATGTTAGATAA**GATAGCTGTGCTAGTAGGCAGTCAGCGCCAT
+r001/1 TTAGATAAAGGATA*CTG
+r002 aaaAGATAA*GGATA
+r003 gcctaAGCTAA
+r004 ATAGCT..............TCAGC
-r003 ttagctTAGGC
-r001/2 CAGCGGCAT
Le format CIGAR (Compact Idiosyncratic Gapped Alignment Report). Il détaille l’alignement d’une lecture sur une séquence de référence. L’alignement est lu de gauche à droite, et retranscrit de manière systématique par une suite de paires (nombre, lettre). Par exemple, le cigar 5M1I5M se décomposen en :
5M : 5 matches entre le fragment et la séquence
1I : une insertion dans le fragment
5M : 5 autres matches.
Soit, par exemple :
read: ACGTAGATCGA
chr1: ACGTA-ATCGA
Les lettres possibles sont :
M : un match (attention, ce peut-être un SNP, mais pas un indel)
I : insertion par rapport à la référence
D : délétion
N : intron
Il existe d’autres lettres, que nous ne détaillerons pas ici.
Autres informations du champ 12 : Il s’agit d’un champ fourre-tout où chaque outil de mapping des informations qu’il juge bon d’ajouter. Ces informations ont un formation particulier, du type : TA:l:valeur, où:
TA est une paire de lettre décrivant le champ
l une autre lettre (pas important)
valeur la valeur du champ.
Voici quelques champs qui peuvent être intéressants :
NM : nombre de mismatches (en comptant les indels) dans l’alignement
AS : score d’alignement
XN: nombre de bases ambigües
XM : nombre de mismatches (sans compter les indels)
XO : nombre d’ouvertures de gaps
X0 : nombre de matches optimaux
X1 : nombre de matches sous-optimaux
MD : description des mismatches
RG : description de l’expérience
YT : description du mapping
UU signifie “single-end”
CP, la paire s’aligne correctement
DP, les deux fragments ne sont pas mappés correctement (en cas d’inversion ou de fusion de gènes)
UP, seul un fragment mappe
SA : autre mapping possible
Il existe beaucoup d’autres tags possibles, qui dépendent des outils de mappings.
See also
Pour en savoir plus Documentation officielle du format: http://samtools.github.io/hts-specs/SAMv1.pdf Article Bioinformatics présentant le format : http://bioinformatics.oxfordjournals.org/content/25/16/2078.long Le format SAM n’est pas destiné à être lu par des programmes. Le format BAM (ci-après) est fait pour cela.
BAM#
Type de fichier#
Mapping
Signification du nom#
Binary sAM
Qui le génère#
Les outils de mapping, les samtools
Qui le lit#
Les visualisateurs, les outils de traitement de lectures
Explications#
Il s’agit simplement du format SAM, binaire (plus facilement lisible pour une machine), et compressé.
BAI#
Type de fichier#
Index
Signification du nom#
BAm Index
Qui le génère#
Les samtools
Qui le lit#
Les visualisateurs
Explications#
Il s’agit d’un fichier binaire qui indexe un fichier BAM. On peut le voir comme une “table des matières”, qui servirait aux outils de visualisation afin d’accélérer l’affichage des données contenues dans un fichier BAM (habituellement très gros).
WIG#
Type de fichier#
Annotation continue sur un génome (comme le GC% ou la densité de lectures)
Signification du nom#
De “wiggle”, qui désigne une course
Qui le génère#
Les outils de peak-calling, entre autres
Qui le lit#
Les visualisateurs
Remarque#
Le format WIG est un format multi-forme. Les différents exemples suivants donneront une densité de lectures sur le génome.
Exemple 1 (variable)#
variableStep chrom=chr2
301 3
302 2
304 2
305 3
Explications#
La première ligne (l’en-tête) décrit le format. variableStep indique que l’annotation sera donnée explicitement pour chaque position du génome. chrom indique le chromosome. Les lignes suivantes indiquent :
(301) la position sur le chromosome
(3) la valeur, ici 3 lectures couvrent la position Si une position n’apparaît pas (dans l’exemple, la position 303), la valeur par défaut est zéro. L’en-tête est répété pour chaque chromosome.
Exemple 2 (variable et span)#
variableStep chrom=chr2 span=5
301 2
306 2.5
Explications#
Dans l’en-tête, le mot-clef span indique que l’information est donnée (moyennée) pour n nucléotides (ici, 5).
Exemple 3 (fixed)#
fixedStep chrom=chr3 start=401 step=100
11
22
33
Explications#
L’en-tête indique que toutes les valeurs sont maintenant données par pas fixés (ici, 100), commençant à la position donnée par start (ici, 401). La valeur 11 correspond donc à un nombre moyen entre les positions 401 et 500, etc. Si le pas (step) n’est pas mentionné, il est par défaut de 1.
See also
Pour en savoir plus Plusieurs sites décrivent en détail ce format :
BedGraph#
Type de fichier#
Annotation continue sur un génome
Signification du nom#
Vient du format BED, destiné à être utilisé en visualisation (“graph”)
Qui le génère#
Les outils de peak-calling, entre autres
Qui le lit#
Les visualisateurs
Exemple#
track type=bedGraph name="BedGraph Format"
chr19 49302000 49302300 -1.0
chr19 49302300 49302600 -0.75
chr19 49302600 49302900 -0.50
chr19 49302900 49303200 -0.25
Explications#
La première ligne (l’en-tête) décrit le format, et le champ “type=bedGraph” est obligatoire. Il existe beaucoup d’autres champs possibles, destinés à l’affichage de l’annotation, non décrits ici. Les lignes suivantes suivent le format BED :
(chr19) le chromosome
(49302000) la position de début
(49302300) la position de fin
(-1.0) la valeur sur l’intervalle
See also
Pour en savoir plus http://genome.ucsc.edu/goldenpath/help/bedgraph.html
BigWig#
Type de fichier#
Annotation continue sur un génome
Signification du nom#
Big Wig
Qui le génère#
Les outils de peak-calling, ou le programme wigtobigwig, bedGraphToBigWig
Qui le lit#
Les visualisateurs
Explications#
Il s’agit d’un fichier binaire qui compresse et indexe un fichier WIG ou BedGraph. Il est donc plus adapté pour des gros fichiers.
Pileup#
Type de fichier#
Variation de séquence
Signification du nom#
“Pile up” signifie “entassement”
Qui le génère#
Les outils de base-calling
Qui le lit#
Les visualisateurs, les outils de génotypage, vous
Exemple#
seq1 272 T 24 ,.$.....,,.,.,...,,,.,..^+. <<<+;<<<<<<<<<<<=<;<;7<&
seq1 273 T 23 ,.....,,.,.,...,,,.,..A <<<;<<<<<<<<<3<=<<<;<<+
seq1 274 T 23 ,.$....,,.,.,...,,,.,... 7<7;<;<<<<<<<<<=<;<;<<6
seq1 275 A 23 ,$....,,.,.,...,,,.,...^l. <+;9*<<<<<<<<<=<<:;<<<<
seq1 276 G 22 ...T,,.,.,...,,,.,.... 33;+<<7=7<<7<&<<1;<<6<
seq1 277 T 22 ....,,.,.,.C.,,,.,..G. +7<;<<<<<<<&<=<<:;<<&<
seq1 278 G 23 ....,,.,.,...,,,.,....^k. %38*<<;<7<<7<=<<<;<<<<<
seq1 279 C 23 A..T,,.,.,...,,,.,..... ;75&<<<<<<<<<=<<<9<<:<<<
Explications#
Chaque ligne contient 5 ou 6 champs:
-(seq1) la référence ou le chromosome
(272) la position sur le chromosome
(T) le nucléotide correspondant à cette position sur le génome de référence
(24) le nombre de lectures à cette position
(,.$…..,,.,.,…,,,.,..^+.) un code correspondant au nucléotide des lectures correspondant à cette position (détaillé ci-après)
(optionnel <<<+;<<<<<<<<<<<=<;<;7<&) la qualité des bases des lectures à cette position (même format que le FASTQ)
Principaux codes :
.(point) : la lecture est sur le brin plus, en accord avec la référence,(virgule) : la lecture est sur le brin moins, en accord avec la référenceACGTN: la lecture est sur le brin plus, en désaccord avec la référence, et contient le nucléotide indiquéacgtn: la lecture est sur le brin moins, en désaccord avec la référence, et contient le nucléotide indiqué+nseq(exemple +2AG) : indique une insertion de n nucléotides, correspondant à la séquence seq (dans l’exemple, insertion de AG).-nseq(exemple -2AG) : indique une suppression de n nucléotides, correspondant à la séquence seq (dans l’exemple, suppression de AG).$le charactère suivant correspond à la fin de la lecture^nle charactère suivant correspond au début de la lecture ; le nombre n indique la qualité du mapping de la lecture (pas de la base)
VCF#
Type de fichier#
Variation de séquence
Signification du nom#
Variant Call Format
Qui le génère#
Les outils de base-calling
Qui le lit#
Les visualisateurs, les outils de génotypage, vous
Exemple#
##fileformat=VCFv4.0
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=AF,Number=.,Type=Float,Description="Allele Frequency">
##FILTER=<ID=q10,Description="Quality below 10">
##FILTER=<ID=s50,Description="Less than 50% of samples have data">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">
##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2
2 10 id1 G A 29 . NS=2;DP=13;AF=0.5 GT:GQ:DP:HQ 0|0:48:1:52,51 1|0:48:8:51,51
2 20 . T A 3 q10 NS=2;DP=12;AF=0.017 GT:GQ:DP:HQ 0|0:46:3:58,50 0|1:3:5:65,3
2 30 id3 A G,T 67 PASS NS=2;DP=10;AF=0.333,0.667 GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2
Explications#
Ce fichier donne des informations sur les variants trouvés dans un génome. Les lignes d’en-tête sont précédées de ##, notamment :
##INFO : Il décrit les informations que l’on collecte pour chaque variant. L’ensemble des informations possible varie avec l’outil utilisé, et ne sera pas décrit ici. Le format est le suivant.
ID (NS) : L’identifiant que l’on retrouvera dans le fichier.
Number (1) : Le nombre de fois que l’on verra l’identifiant pour chaque variant (le point signifie “un nombre arbitraire”).
Type (Integer) : Le type d’information, comme un entier, un mot, etc.
Description (Number of Samples with Data) : Une description de l’information.
##FILTER : L’ensemble des étapes de filtrage utilisées pour générer ce fichier.
ID (q10) : Identifiant du filtrage.
Description (Quality below 10) : Description du filtrage.
##FORMAT : Données du génotypage trouvées pour chaque variant.
ID (GQ) : Identifiant du génotypage.
Number (1) : Le nombre de fois que l’on verra cette donnée pour chaque variant.
Type (Integer) : Le type d’information, comme un entier, un mot, etc.
Description (Genotype Quality) : Une description du génotypage.
Chaque ligne contient au moins 10 champs. Les champs 10 et plus décrivent les génotypages dans les différents échantillons.
(2) le chromosome
(10) la position sur le chromosome
(id1) l’identifiant du variant
(G) variant sur la référence
(A) variant sur les lectures
(29) score de qualité sur le variant (score Phred)
(.) filtres passés : PASS si tous les filtres sont passés ; un point si aucun n’est passé ; sinon on énumère la liste des filtres qui sont passés (tels que définis par les en-tête ##FILTER), séparés par un point-virgule.
(NS=2;DP=13;AF=0.5) Informations sur le variant, qui utilise la nomenclature définie dans les en-tête ##INFO. Ici, il s’agit qu’un variant que l’on observe dans les 2 échantillons, avec une profondeur de 13 lectures, et une fréquence de 50%.
(GT:GQ:DP:HQ) Format des données de génotypages pour chaque échantillon, séparés par des “deux-points”.
(0|0:48:1:52,51) Données de génotypage pour l’échantillon 1. Ici, nous avons le génotypage (GG), qualité du génotypage (score Phred), profondeur de séquençage (une lecture), qualité des haplotypes (un score Phred par haplotype).
et suivant Données de génotypage pour l’échantillon n.
See also
Pour en savoir plus Définition du format: https://samtools.github.io/hts-specs/VCFv4.2.pdf