Redirections#
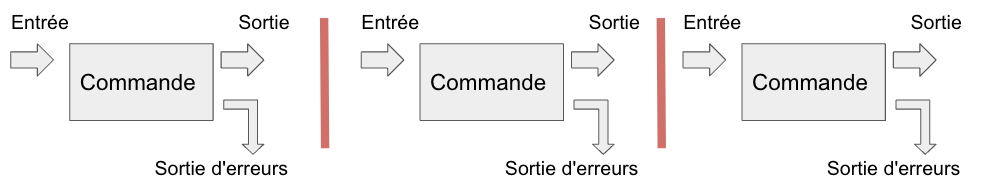
Entrée standard (stdin): un fichier ou du texte (un flux de texte).
Sortie standard (stdout, “output 1”): renvoyée à l’écran par défaut et transférée via le tube.
Erreur standard (stderr, “output 2”): renvoyée à l’écran par défaut et non transférée via le tube.
Opérateurs de redirection
|: le caractère “pipe” passe le flux de texte stdin à une autre commande> file.txt: stocke le flux stdout en créant (ou écrasant) le fichier file.txt>> file.txt: stocke le flux stdout en ajoutant des lignes dans le fichier file.txt2> log.txt: stocke le flux stderr dans un fichier nommé log.txt1> file.txt 2> log.txt: stocke stdout dans un fichier et stderr dans un autre
Note
Si vous souhaitez rediriger stdout et stderr dans le même fichier : &> monFichier.txt
Un exemple#
Compter le nombre de fichiers d’extension “bed” présents dans le répertoire où vous êtes.
cd /shared/projects/form_2022_32/coursLinux/demo/annotations/hg38/bed
ls -l *.bed | wc -l
1
?2004h
Exercices#
Hommage à Marseille
Utilisez les commandes head pour visualiser les 51 premières lignes du fichier hg38_exons.bed et renvoyer le résultat dans less
Hommage au finistère
Utilisez les commandes head et tail pour récupérer la 29ème ligne du fichier hg38_exons.bed.
# On utilise head puis un tube qui renvoie vers less.
# Tapez q pour quitter.
head -n 51 hg38_exons.bed | less
# On récupère les 29 premières lignes avec head,
# puis on extrait de ce flux de texte la dernière ligne avec tail
head -n 29 hg38_exons.bed | tail -n 1
Demo: enchaînements de commandes#
# Obtenir la liste non redondante de chromosomes présents dans le fichier
cut -f 1 hg38_exons.bed | sort | uniq
chr1
chr10
chr11
chr12
chr13
chr14
chr15
chr16
chr17
chr18
chr19
chr2
chr20
chr21
chr22
chr3
chr4
chr5
chr6
chr7
chr8
chr9
chrX
chrY
# Nombre de chromosomes différents
cut -f 1 hg38_exons.bed | sort | uniq | wc -l
24
# Obtenir la liste des chromosomes présents dans le fichier et
# le nombre d'occurrence de chacun d'entre eux
# ‘-c=count’
cut -f 1 hg38_exons.bed | sort | uniq -c
117347 chr1
46414 chr10
75294 chr11
72970 chr12
20371 chr13
43618 chr14
47068 chr15
60375 chr16
77950 chr17
22378 chr18
73716 chr19
97092 chr2
28097 chr20
14332 chr21
27854 chr22
80596 chr3
49351 chr4
55719 chr5
56433 chr6
60961 chr7
45192 chr8
45468 chr9
39510 chrX
3764 chrY
# La liste des chromosomes présents dans le fichier et leur nombre trié
# par ordre décroissant (-r: reverse, -n: numeric, -k: ‘kolonne’)
cut -f 1 hg38_exons.bed | sort | uniq -c | sort -nr -k 1,1
117347 chr1
97092 chr2
80596 chr3
77950 chr17
75294 chr11
73716 chr19
72970 chr12
60961 chr7
60375 chr16
56433 chr6
55719 chr5
49351 chr4
47068 chr15
46414 chr10
45468 chr9
45192 chr8
43618 chr14
39510 chrX
28097 chr20
27854 chr22
22378 chr18
20371 chr13
14332 chr21
3764 chrY
Note
La commande uniq permet d’éliminer les doublons dans un flux de texte trié
Exercice I#
En quoi la commande
lessdiffère-t-elle de la commandemore? (15 pts)En utilisant la command
grepindiquez : (5 pts)Combien y-a-t-il d’exons sur le chromosome 22 ?
Combien de lignes correspondant aux exons présents sur le chromosome chr22 contiennent le terme lincRNA ?
Solution#
grep "chr22" hg38_exons.bed | wc -l
27854
grep "chr22" hg38_exons.bed | grep "lincRNA" | wc -l
846
grep -c "chr22" hg38_exons.bed
27854
grep "chr22" hg38_exons.bed | grep -c "lincRNA"
846