EBAII
Supports de cours de l'Ecole de Bioinformatique Aviesan - IFB - Inserm "Initiation au traitement des données de génomique obtenues par séquençage à haut débit"
ChIP-seq Hands-on Roscoff 2021
- Introduction
- Downloading ChIP-seq reads from NCBI
- Connect to the server and set up your environment
- Quality control of the reads and statistics
- Mapping the reads with Bowtie
- Estimating the number of duplicated reads
- ChIP quality controls
- Visualizing the data in a genome browser
- Peak calling with MACS
- Motif analysis
- Peak annotation
- Bonus: Peak annotation using R
- FAQ
- References
Introduction
Goal
The aim is to :
- understand the nature of ChIP-Seq data
- perform a complete analysis workflow including quality check (QC), read mapping, visualization in a genome browser and peak-calling. Use command line and open source software for each step of the workflow and feel the complexity of the task
- have an overview of some possible downstream analyses
- perform a motif analysis with online web programs
Summary
This training gives an introduction to ChIP-seq data analysis, covering the processing steps starting from the reads to the peaks. Among all possible downstream analyses, the practical aspect will focus on motif analyses. A particular emphasis will be put on deciding which downstream analyses to perform depending on the biological question. This training does not cover all methods available today. It does not aim at bringing users to a professional NGS analyst level but provides enough information to allow biologists understand what is DNA sequencing in practice and to communicate with NGS experts for more in-depth needs.
Dataset description
For this training, we will use two datasets:
- a dataset produced by Myers et al Pubmed involved in the regulation of gene expression under anaerobic conditions in bacteria. We will focus on one factor: FNR. The advantage of this dataset is its small size, allowing real time execution of all steps of the dataset
- a dataset of ChIP-seq peaks obtained in different mouse tissues for the p300 co-activator protein by Visel et al. Pubmed; we will use this dataset to illustrate downstream annotation of peaks using R.
Downloading ChIP-seq reads from NCBI
Goal: Identify the datasets corresponding to the studied article and retrieve the data (reads as FASTQ files) corresponding to 2 replicates of a condition and the corresponding control.
1 - Obtaining an identifier for a chosen dataset
NGS datasets are (usually) made freely accessible for other scientists, by depositing these datasets into specialized databanks. Sequence Read Archive (SRA) located in USA hosted by NCBI, and its European equivalent European Nucleotide Archive (ENA) located in England hosted by EBI both contains raw reads.
Functional genomic datasets (transcriptomics, genome-wide binding such as ChIP-seq,…) are deposited in the databases Gene Expression Omnibus (GEO) or its European equivalent ArrayExpress.
Within an article of interest, search for a sentence mentioning the deposition of the data in a database. Here, the following sentence can be found at the end of the Materials and Methods section: “All genome-wide data from this publication have been deposited in NCBI’s Gene Expression Omnibus (GSE41195).” We will thus use the GSE41195 identifier to retrieve the dataset from the NCBI GEO (Gene Expression Omnibus) database.
2 - Accessing GSE41195 from GEO
- The GEO database hosts processed data files and many details related to the experiments. SRA (Sequence Read Archive) stores the actual raw sequence data.
- Search in Google GSE41195. Click on the first link to directly access the correct page on the GEO database.
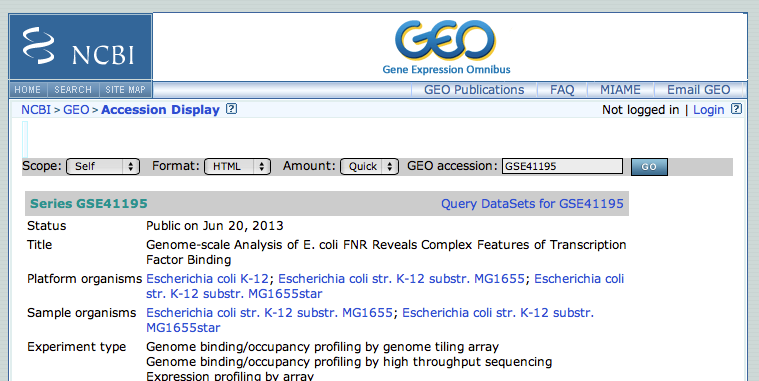
- This GEO entry is a mixture of expression analysis (Nimblegen Gene Expression Array), chip-chip and chip-seq. At the bottom of the page, click on the subseries related to the chip-seq datasets. (this subseries has its own identifier: GSE41187).

- From this page, we will focus on the experiment FNR IP ChIP-seq Anaerobic A. At the bottom of the page, click on the link “GSM1010219 - FNR IP ChIP-seq Anaerobic A”.
- In the new page, go to the bottom to find the SRA identifier. This is the identifier of the raw dataset stored in the SRA database.
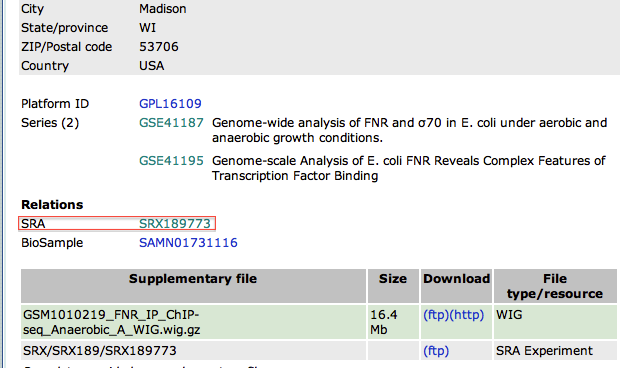
- Copy the identifier SRX189773 (do not click on the link that would take you to the SRA database, see below why)
3 - Downloading FASTQ file from the ENA database
Although direct access to the SRA database at the NCBI is doable, SRA does not store sequences in a FASTQ format. So, in practice, it’s simpler (and quicker!!) to download datasets from the ENA database (European Nucleotide Archive) hosted by EBI (European Bioinformatics Institute) in UK. ENA encompasses the data from SRA.
- Go to the EBI website. Paste your SRA identifier (SRX189773) and click on the button “search”.
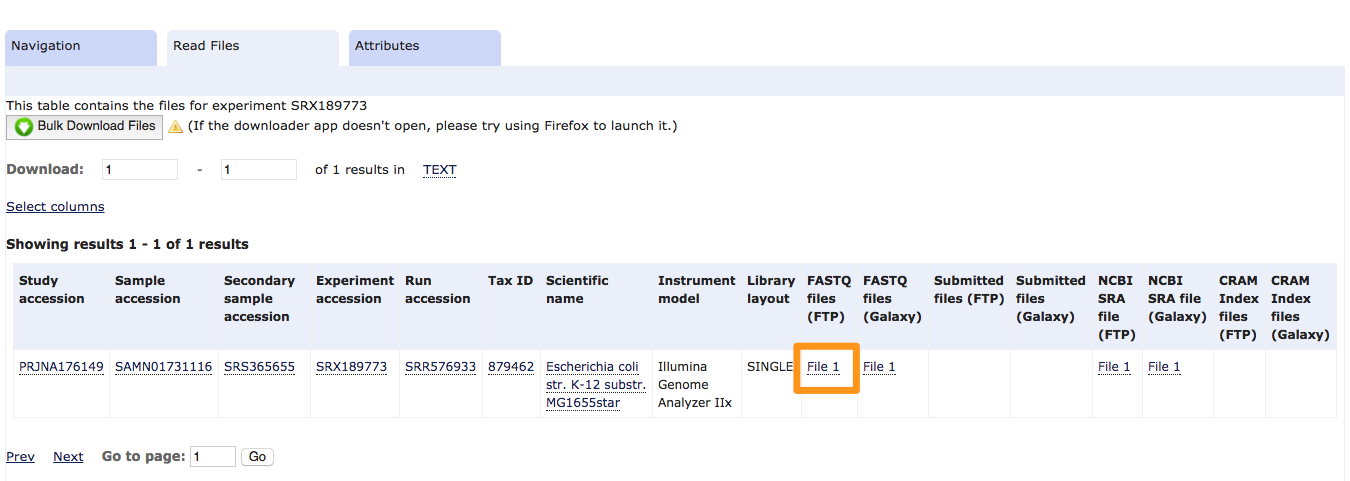
- Click on the first result. On the next page, there is a link to the FASTQ file. For efficiency, this file has already been downloaded and is available in the “data” folder (SRR576933.fastq.gz)
tip: To download the replicate and control datasets, we should redo the same steps starting from the GEO web page specific to the chip-seq datasets (see step 2.4) and choose FNR IP ChIP-seq Anaerobic B and anaerobic INPUT DNA. Downloaded FASTQ files are available in the data folder (SRR576934.fastq.gz and SRR576938.fastq.gz respectively)
At this point, you have three FASTQ files, two IPs, one control (INPUT).
Connect to the server and set up your environment
1 - Sign in to Jupyterhub and open a Terminal
- Go to Jupyterhub
- select a job profile: EBAII and click on “Start”
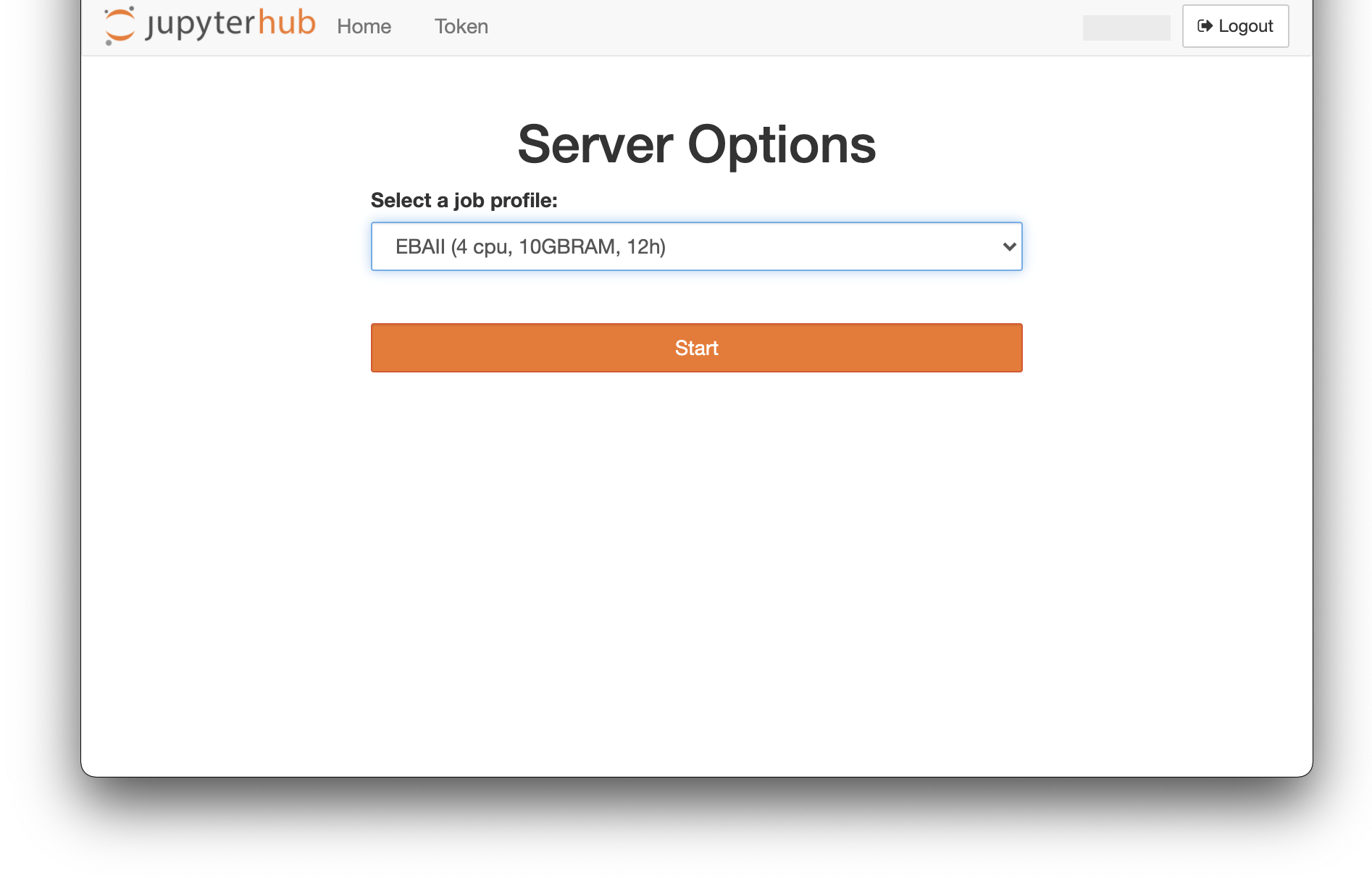
- In the launcher, click on “Terminal” in “Other” section. You should be in your home directory by default. Check it:
pwd
2 - Set up your working environment
- Go to your project directory
cd /shared/projects/<your_project> - Create a directory that will contain all results of the upcoming analyses.
mkdir EBAII2021_chipseq - Go to the newly created directory
cd EBAII2021_chipseq - Copy the directory containing data
cp -r /shared/home/slegras/EBAII2021_chipseq/data .
- Your directory structure should be like this
/shared/projects/<your_project>/EBAII2021_chipseq │ └───data
If you wish, you can check your directory structure:
tree
Quality control of the reads and statistics
Goal: Get some basic information on the data (read length, number of reads, global quality of datasets)
1 - Generating the FASTQC report
Before you analyze the data, it is crucial to check the quality of the data. We will use the standard tool for checking the quality of data generated on the Illumina platform: FASTQC.
- Create a directory named 01-QualityControl in which to output results from fastqc
mkdir 01-QualityControl - Go to the directory you’ve just created
cd 01-QualityControl
Your directory structure should be like this
/shared/projects/<your_project>/EBAII2021_chipseq
│
└───data
│
└───01-QualityControl <- you should be here
- Get FastQC available in your environment
module add fastqc/0.11.9 - Check the help page of the program to see its usage and parameters.
fastqc --help
- Launch the FASTQC program on the experiment file (SRR576933.fastq.gz)
- -o: creates all output files in the specified output directory. ‘.’ means current directory.
fastqc ../data/SRR576933.fastq.gz -o .
- -o: creates all output files in the specified output directory. ‘.’ means current directory.
- Wait until the analysis is finished. Check the FastQC result files.
lsSRR576933_fastqc.html SRR576933_fastqc.zip
-
Go to the directory /shared/projects/
/EBAII2021_chipseq/1-QualityControl in the tree diretory on the left of the jupyterhub panel and double click on SRR576933_fastqc.html to visualize the file.  - Launch the FASTQC program on the replicate (SRR576934.fastq.gz) and on the control file (SRR576938.fastq.gz)
Analyze the result of the FASTQC program:
How many reads are present in each file ?
What is the read length ?
Is the overall quality good for the three samples ?
Are there any concerns raised by the report ? If so, can you tell where the problem might come from ?
- Once you are done with FastQC, unload it
module rm fastqc/0.11.9
Mapping the reads with Bowtie
Goal: Obtain the coordinates of each read to the reference genome.
1 - Choosing a mapping program
There are multiple programs to perform the mapping step. For reads produced by an Illumina machine for ChIP-seq, the currently “standard” programs are BWA (Li and Durbin, 2009) and Bowtie (versions 1 and 2)(Langmead et al., 2009; Langmead and Salzberg, 2012), and STAR (Dobin et al., 2013) is getting popular. We will use Bowtie version 1.2.1.1 for this exercise, as this program remains effective for short reads (< 50bp).
2 - Bowtie
- Load Bowtie
module add bowtie/1.2.3 - Try out bowtie
bowtieThis prints the help of the program. However, this is a bit difficult to read ! If you need to know more about the program, it’s easier to directly check out the manual on the website.
-
bowtie needs the reference genome to align each read on it. This genome needs to be in a specific format (=index) for bowtie to be able to use it. Several pre-built indexes are available for download on bowtie webpage, but our genome is not there. You will need to make this index file.
- Create a directory named 02-Mapping in which to output mapping results
cd .. mkdir 02-Mapping - Go to the directory you’ve just created
cd 02-Mapping
3 - Prepare the index file
-
To make the index file, you will need the complete genome, in FASTA format. It has already been downloaded to gain time (Escherichia_coli_K12.fasta in the course folder) (The genome was downloaded from the NCBI).
- Create a directory named index in which to output bowtie indexes
mkdir index - Go to the newly created directory
cd index - Try out bowtie-build
bowtie-build - Build the index for bowtie
## Creating genome index : provide the path to the genome file and the name to give to the index (Escherichia_coli_K12) bowtie-build ../../data/Escherichia_coli_K12.fasta Escherichia_coli_K12 - Go back to upper directory i.e 02-Mapping
cd ..
4 - Mapping the IP samples
- Create a directory named IP to put mapping results for IP
mkdir bam - Go to the newly created directory bam
cd bamYour directory structure should be like this:
/shared/projects/<your_project>/EBAII2021_chipseq │ └───data │ └───01-QualityControl │ └───02-Mapping | └───index | └───bam <- you should be here - Let’s see the parameters of bowtie before launching the mapping:
- Escherichia_coli_K12 is the name of our genome index file
- Number of mismatches for SOAP-like alignment policy (-v): to 2, which will allow two mismatches anywhere in the read, when aligning the read to the genome sequence.
- Suppress all alignments for a read if more than n reportable alignments exist (-m): to 1, which will exclude the reads that do not map uniquely to the genome.
- -q indicates the input file is in FASTQ format. SRR576933.fastq is the name of our FASTQ file.
- -3 will trim x base from the end of the read. As our last position is of low quality, we’ll trim 1 base.
- -S will output the result in SAM format
- 2> SRR576933.out will output some statistics about the mapping in the file SRR576933.out
## Run alignment ## Tip: first type bowtie command line then add quotes around and prefix it with "sbatch --cpus 10 --wrap=" sbatch --cpus-per-task 10 --wrap="bowtie -p 10 ../../index/Escherichia_coli_K12 ../../../data/SRR576933.fastq.gz -v 2 -m 1 -3 1 -S 2> SRR576933.out > SRR576933.sam"This should take few minutes as we work with a small genome. For the human genome, we would need either more time and more resources.
Bowtie output is a SAM file. The SAM format corresponds to large text files, that can be compressed (“zipped”) into a BAM format. The BAM files takes up to 4 time less disk space and are usually sorted and indexed for fast access to the data it contains. The index of a given
- Sort the sam file and create a bam file using samtools
- -b: output BAM
## First load samtools module add samtools/1.10 ## Then run samtools samtools sort SRR576933.sam | samtools view -b > SRR576933.bam
- -b: output BAM
- Create an index for the bam file
samtools index SRR576933.bam - Compress the .sam file (you could also delete the file)
gzip SRR576933.sam
Analyze the result of the mapped reads:
Open the file SRR576933.out (for example using the ` less ` command), which contains some statistics about the mapping. How many reads were mapped? How many multi-mapped reads were originally present in the sample? To quit less press ‘q’
- Once it’s done, unload the tools you used
module rm samtools/1.10 bowtie/1.2.3
5 - Map the second replicate and the control
- Repeat the steps above (in 4 - Mapping the experiment) for the files SRR576934.fastq.gz and SRR576938.fastq.gz in the directory named “bam” within the directory 02-Mapping.
Analyze the result of the mapped reads:
Open the file SRR576938.out. How many reads were mapped?
Estimating the number of duplicated reads
Goal: Duplicated reads i.e reads mapped at the same positions in the genome are present in ChIP-seq results. They can arise for several reasons including a biased amplification during the PCR step of the library prep, DNA fragment coming from repetitive elements of the genome, sequencing saturation or the same clusters read several times on the flowcell (i.e optical duplicates). As analyzing ChIP-Seq data consist at some point in detecting signal enrichment, we can not keep duplicated reads for subsequent analysis. So let’s detect them using Picard
- Go to the directory with alignment files
cd /shared/projects/<your_project>/EBAII2021_chipseq/02-Mapping/bam - Run Picard markDuplicates to mark duplicated reads (= reads mapping at the exact same location on the genome)
- CREATE_INDEX: Create .bai file for the result bam file with marked duplicate reads
- INPUT: input file name to mark for duplicate reads
- OUTPUT: output file name
- METRICS: file with duplicates marking statistics
- VALIDATION_STRINGENCY: Validation stringency for all SAM files read by picard.
## Load picard
module add picard/2.22.0
## Run picard
picard MarkDuplicates \
CREATE_INDEX=true \
INPUT=SRR576933.bam \
OUTPUT=Marked_SRR576933.bam \
METRICS_FILE=metrics \
VALIDATION_STRINGENCY=STRICT
picard MarkDuplicates \
CREATE_INDEX=true \
INPUT=SRR576938.bam \
OUTPUT=Marked_SRR576938.bam \
METRICS_FILE=metrics \
VALIDATION_STRINGENCY=STRICT
To determine the number of duplicated reads marked by Picard, we can run the samtools flagstat command:
## Add samtools to your environment
module add samtools/1.10
## run samtools
samtools flagstat Marked_SRR576933.bam
Go back to working home directory (i.e /shared/projects/
## Unload picard and samtools
module rm samtools/1.10 picard/2.22.0
## If you are in 02-Mapping/bam
cd ../..
ChIP quality controls
Goal: The first exercise aims at plotting the Lorenz curve to assess the quality of the chIP. The second exercise aims at calculating the NSC and RSC ENCODE quality metrics. These metrics allow to classify the datasets (after mapping, contrary to FASTQC that works on raw reads) in regards to the NSC and RSC values observed in the ENCODE datasets (see ENCODE guidelines)
1 - Plot the Lorenz curve with Deeptools
- Create a directory named 03-ChIPQualityControls in which to put mapping results for IP
mkdir 03-ChIPQualityControls - Go to the newly created directory
cd 03-ChIPQualityControls - Run Deeptools plotFingerprint (Fidel et al, NAR, 2016) to draw the Lorenz curve
- -b: List of indexed BAM files
- -plot: File name of the output figure (extension can be either “png”, “eps”, “pdf” or “svg”)
## Load deeptools in your environment module add deeptools/3.2.0 ## Run deeptools fingerprint plotFingerprint --numberOfSamples 10000 -b ../02-Mapping/bam/SRR576933.bam ../02-Mapping/bam/SRR576934.bam ../02-Mapping/bam/SRR576938.bam -plot fingerprint_10000.png- If plotFingerprint takes ages to run. Take the file that has already been prepared for the training.
cp /shared/home/slegras/EBAII2021_chipseq/03-ChIPQualityControls/fingerprint.png . - Go to the file using the directory tree on the left of the Jupyterhub panel and click on the fingerprint.png file to display it in Jupyterhub.
- If plotFingerprint takes ages to run. Take the file that has already been prepared for the training.
Look at the result files fingerprint.png. What do you think of it?
Go back to working home directory (i.e /shared/projects/training/<login>/EBAII2021_chipseq)
## Unload deepTools
module rm deeptools/3.2.0
## If you are in 03-ChIPQualityControls
cd ..
Visualizing the data in a genome browser
Goal: View the data in their genomic context, to check whether the IP worked
1 - Choosing a genome browser
There are several options for genome browsers, divided between the local browsers (need to install the program, eg. IGV) and the online web browsers (eg. UCSC genome browser, Ensembl). We often use both types, depending on the aim and the localization of the data. If the data are on your computer, to prevent data transfer, it’s easier to visualize the data locally (IGV). Note that if you’re working on a non-model organism, the local viewer will be the only choice. If the aim is to share the results with your collaborators, view many tracks in the context of many existing annotations, then the online genome browsers are more suitable.
2 - Viewing the raw alignment data in IGV
- Download the following files from the server onto your computer
- data/Escherichia_coli_K12.fasta
- data/Escherichia_coli_K_12_MG1655.annotation.fixed.gtf.gz
- 02-Mapping/bam/SRR576933.bam
- 02-Mapping/bam/SRR576933.bam.bai
- 02-Mapping/bam/SRR576934.bam
- 02-Mapping/bam/SRR576934.bam.bai
- 02-Mapping/bam/SRR576938.bam
- 02-Mapping/bam/SRR576938.bam.bai
- Open IGV on your computer
- Load the genome
- Genomes / Load Genome from File…
- Select the fasta file Escherichia_coli_K12.fasta located into the data directory
- Load an annotation file named Escherichia_coli_K12_MG1655.annotation.gff3 into IGV
- File / Load from File…
- Select the annotation file. The positions of the genes are now loaded.
- Load the three bam files (SRR576933.bam, SRR576934.bam and SRR576938.bam) in IGV.
Browse around in the genome. Do you see peaks?
Browse into IGV. Go to the following genes: pepT (geneID:b1127), ycfP (geneID:b1108)
However, looking at BAM file as such does not allow to directly compare the two samples as data are not normalized. Let’s generate normalized data for visualization.
3 - Viewing scaled data
bamCoverage from deepTools generates BigWigs out of BAM files
- Try it out
## Load deeptools in your environment module add deeptools/3.2.0 ## run bamCoverage bamCoverage --help - Create a directory named 04-Visualization to store bamCoverage outputs
mkdir 04-Visualization - Go to the newly created directory
cd 04-Visualization
Your directory structure should be like this:
/shared/projects/<your_project>/EBAII2021_chipseq
│
└───data
│
└───01-QualityControl
│
└───02-Mapping
| └───index
| └───bam
│
└───03-ChIPQualityControls
│
└───04-Visualization <- you should be in this folder
- Generate a scaled bigwig file on the IP with bamCoverage
- –bam: BAM file to process
- –outFileName: output file name
- –outFileFormat: output file type
- –effectiveGenomeSize : size of the mappable genome
- –normalizeUsing : different overall normalization methods; we will use RPGC method corresponding to 1x average coverage
- –skipNonCoveredRegions: skip non-covered regions
- –extendReads 200: Extend reads to fragment size
- –ignoreDuplicates: reads that have the same orientation and start position will be considered only once
bamCoverage --bam ../02-Mapping/bam/Marked_SRR576933.bam \ --outFileName SRR576933_nodup.bw --outFileFormat bigwig --effectiveGenomeSize 4639675 \ --normalizeUsing RPGC --skipNonCoveredRegions --extendReads 200 --ignoreDuplicates
- Do it for the replicate and the control.
- Download the three bigwig files you have just generated
- 04-Visualization/SRR576933_nodup.bw
- 04-Visualization/SRR576934_nodup.bw
- 04-Visualization/SRR576938_nodup.bw
- Load the three bigwig files in IGV
- File / Load from File…
- Select the three bigwig files.
- Set the visualization of the three bigwig files to be autoscaled
- Click right on the name of the tracks and select Autoscale
Go back to the genes we looked at earlier: pepT, ycfP. Look at the shape of the signal.
Keep IGV opened.
Go back to working home directory (i.e /shared/projects/
## If you are in 04-Visualization
cd ..
Peak calling with MACS2
Goal: Define the peaks, i.e. the region with a high density of reads, where the studied factor was bound
1 - Choosing a peak-calling program
There are multiple programs to perform the peak-calling step. Some are more directed towards histone marks (broad peaks) while others are specific to narrow peaks (transcription factors). Here we will use the callpeak function of MACS2 (version 2.1.1.20160309) because it’s known to produce generally good results, and it is well-maintained by the developer.
2 - Calling the peaks
- Create a directory named 05-PeakCalling and one directory named replicates within to store peaks coordinates.
mkdir 05-PeakCalling mkdir 05-PeakCalling/replicates - Go to the newly created directory replicates
cd 05-PeakCalling/replicates - Try out MACS2
## Load macs2 in your environment module add macs2/2.2.7.1 macs2 callpeakThis prints the help of the program.
- Let’s see the parameters of MACS before launching the mapping:
- ChIP-seq tag file (-t) is the name of our experiment (treatment) mapped read file SRR576933.bam
- ChIP-seq control file (-c) is the name of our input (control) mapped read file SRR576938.bam
- –format BAM indicates the input file are in BAM format. Other formats can be specified (SAM,BED…)
- –gsize Effective genome size: this is the size of the genome considered “usable” for peak calling. This value is given by the MACS developers on their website. It is smaller than the complete genome because many regions are excluded (telomeres, highly repeated regions…). The default value is for human (2700000000.0), so we need to change it. As the value for E. coli is not provided, we will take the complete genome size 4639675.
- –name provides a prefix for the output files. We set this to FNR_Anaerobic_A, but it could be any name.
- –bw The bandwidth is the size of the fragment extracted from the gel electrophoresis or expected from sonication. By default, this value is 300bp. Usually, this value is indicated in the Methods section of publications. In the studied publication, a sentence mentions “400bp fragments (FNR libraries)”. We thus set this value to 400.
- –keep-dup specifies how MACS should treat the reads that are located at the exact same location (duplicates). The manual specifies that keeping only 1 representative of these “stacks” of reads is giving the best results. We doesn’t mention it as 1 is the default value.
- –fix-bimodal indicates that in the case where macs2 cannot find enough paired peaks between the plus strand and minus strand to build the shifting model, it can bypass this step and use a extension size of 200bp by default.
- -p 1e-2 indicates that we report the peaks if their associated p-value is lower than 1e-2. This is a relaxed threshold as we want to keep a high number of false positives in our peak set to later compute the IDR analysis.
- &> MACS.out will output the verbosity (=information) in the file MACS.out
macs2 callpeak -t ../../02-Mapping/bam/SRR576933.bam \ -c ../../02-Mapping/bam/SRR576938.bam --format BAM \ --gsize 4639675 --name 'FNR_Anaerobic_A' --bw 400 \ --fix-bimodal -p 1e-2 &> repA_MACS.out- Run macs2 for replicate A, then go to repB directory and run macs2 for replicate B by changing the treatment file (-t), the output file name (-n) and the log file (&>), this should take a few minutes each.
- In a new directory called pool, run macs2 for the pooled replicates A and B by giving both bam files as input treatment files (-t).
# If you are in 05-PeakCalling
mkdir pool
cd pool
# Run macs2 for pooled replicates
macs2 callpeak -t ../../02-Mapping/bam/SRR576933.bam ../../02-Mapping/bam/SRR576934.bam \
-c ../../02-Mapping/bam/SRR576938.bam --format BAM \
--gsize 4639675 --name 'FNR_Anaerobic_pool' --bw 400 \
--fix-bimodal -p 1e-2 &> pool_MACS.out
3 - Analyzing the MACS results
Look at the files that were created by MACS. Which files contains which information ?
How many peaks were detected by MACS ?
At this point, you should have two BED files containing the peak coordinates, one for each replicate.
4 - Combine replicate together (IDR)
In order to take advantage of having biological replicates, we will create a combine set of peaks based on the reproducibility of each individual replicate peak calling. We will use the Irreproducible Discovery Rate (IDR) algorithm.
- Create a new directory to store the peak coordinates resulting after idr analysis
## If you are in 05-PeakCalling mkdir idr cd idrYour directory structure should be like this:
/shared/projects/<your_project>/EBAII2021_chipseq │ └───data │ └───01-QualityControl │ └───02-Mapping | └───index | └───bam │ └───03-ChIPQualityControls │ └───04-Visualization | └───05-PeakCalling | └───replicates | └───pool | └───idr <- you should be in this folder - Load the module idr and have a look at its parameters
## Load idr in your environment module add idr/2.0.4.2 idr --help
- –samples : peak files of each individual replicate
- –peak-list : the peak file of the pooled replicates, it will be used as a master peak set to compare with the regions from each replicates
- –input-file-type : format of the peak file, in our case it is narrowPeak
- –output-file : name of the result file
- –plot : plot additional diagnosis plot
- Run idr
idr --samples ../replicates/FNR_Anaerobic_A_peaks.narrowPeak ../replicates/FNR_Anaerobic_B_peaks.narrowPeak \ --peak-list ../pool/FNR_Anaerobic_pool_peaks.narrowPeak \ --input-file-type narrowPeak --output-file FNR_anaerobic_idr_peaks.bed \ --plot - Remove IDR and MACS2 from your environment and go back to working home directory (i.e /shared/projects/
/EBAII2021_chipseq) ```bash module rm macs2/2.1.1.20160309 module rm idr/2.0.4.2
If you are in 05-PeakCalling/idr
cd ../..
### 5 - Visualize peaks into IGV
1. Download the following BED files from the server into your computer to visualise in IGV :
* 05-PeakCalling/repA/FNR_Anaerobic_A_peaks.bed
* 05-PeakCalling/repB/FNR_Anaerobic_B_peaks.bed
* 05-PeakCalling/idr/FNR_anaerobic_idr_peaks.bed
**Go back again to the genes we looked at earlier: pepT, ycfP. Do you see peaks?**
## Motif analysis <a name="motif"></a>
**Goal**: Define binding motif(s) for the ChIPed transcription factor and identify potential cofactors
### 1 - Retrieve the peak sequences corresponding to the peak coordinate file (BED)
For the motif analysis, you first need to extract the sequences corresponding to the peaks. There are several ways to do this (as usual...). If you work on a UCSC-supported organism, the easiest is to use RSAT fetch-sequences or Galaxy. Here, we will use Bedtools, as we have the genome of interest on our computer (Escherichia_coli_K12.fasta).
1. Create a directory named **06-MotifAnalysis** to store data needed for motif analysis
```bash
mkdir 06-MotifAnalysis
- Go to the newly created directory
cd 06-MotifAnalysis
Your directory structure should be like this:
/shared/projects/<your_project>/EBAII2021_chipseq
│
└───data
│
└───01-QualityControl
│
└───02-Mapping
| └───index
| └───bam
│
└───03-ChIPQualityControls
│
└───04-Visualization
│
└───05-PeakCalling
│
└───06-MotifAnalysis <- you should be in this folder
- Extract peak sequence in fasta format
```bash
First load samtools
module add samtools/1.10
Create an index of the genome fasta file
samtools faidx ../data/Escherichia_coli_K12.fasta
First load bedtools
module add bedtools/2.29.2
Extract fasta sequence from genomic coordinate of peaks
bedtools getfasta -fi ../data/Escherichia_coli_K12.fasta
-bed ../05-PeakCalling/idr/FNR_anaerobic_idr_peaks.bed -fo FNR_anaerobic_idr_peaks.fa
4. Download the file FNR_anaerobic_idr_peaks.fa on your computer
### 2 - Motif discovery with RSAT
1. Open a connection to a Regulatory Sequence Analysis Tools server. You can choose between various website mirrors.
* Teaching Server (recommended for this training) [http://pedagogix-tagc.univ-mrs.fr/rsat/](http://pedagogix-tagc.univ-mrs.fr/rsat/)
2. In the left menu, click on **NGS ChIP-seq** and then click on **peak-motifs**. A new page opens, with a form
3. The default peak-motifs web form only displays the essential options. There are only two mandatory parameters.
* The **title box**, which you will set as **FNR Anaerobic** . The **sequences**, that you will **upload from your computer**, by clicking on the button Choose file, and select the file **FNR_anaerobic_idr_peaks.fa** from your computer.
4. We will now modify some of the advanced options in order to fine-tune the analysis according to your data set.
* Open the "Reduce peak sequences" title, and make sure the **Cut peak sequences: +/- ** option is set to **0** (we wish to analyze our full dataset)
* Open the “Motif Discovery parameters” title, and check the **oligomer sizes 6 and 7** (but not 8). Check "Discover over-represented spaced word pairs **[dyad-analysis]**"
* Under “Compare discovered motifs with databases”, **remove** "JASPAR core vertebrates" and **add RegulonDB prokaryotes** (2015_08) as the studied organism is the bacteria E. coli.
5. You can indicate your email address in order to receive notification of the task submission and completion. This is particularly useful because the full analysis may take some time for very large datasets.
6. Click “**GO**”. As soon as the query has been launched, you should receive an email indicating confirming the task submission, and providing a link to the future result page.
7. The Web page also displays a link, You can already click on this link. The report will be progressively updated during the processing of the workflow.
### 3 - OPTIONAL : Motif discovery with RSAT (short peaks)
1. Restrict the dataset to the summit of the peaks +/- 100bp using bedtools slop. Using bedtools slop to extend genomic coordinates allow not to go beyond chromosome boundaries as the user give the size of chromosomes as input (see fai file).
```bash
bedtools slop -b 100 -i ../05-PeakCalling/replicates/FNR_Anaerobic_A_summits.bed -g ../data/Escherichia_coli_K12.fasta.fai > FNR_Anaerobic_A_summits+-100.bed
- Extract the sequences for this BED file
```bash
Extract fasta sequence from genomic coordinate of peaks
bedtools getfasta -fi ../data/Escherichia_coli_K12.fasta -bed FNR_Anaerobic_A_summits+-100.bed -fo FNR_Anaerobic_A_summits+-100.fa
Compress the genome file as we won’t need it anymore
gzip ../data/Escherichia_coli_K12.fasta
3. Run RSAT peak-motifs with the same options, but choosing as input file this new dataset (FNR_Anaerobic_A_summits+-100.fa)
and setting the title box to **FNR Anaerobic A summit +/-100bp**
## Peak annotation <a name="annotation"></a>
**Goals**: Associate ChIP-seq peaks to genomic features, draw metagenes, identify closest genes and run ontology analyses
1. Create a directory named **07-PeakAnnotation**
```bash
mkdir 07-PeakAnnotation
- Go to the newly created directory
cd 07-PeakAnnotation
1-Associate peaks to closest genes
annotatePeaks.pl from the Homer suite associates peaks with nearby genes.
- We will need the annotation file data/Escherichia_coli_K_12_MG1655.annotation.fixed.gtf.gz and the genome file data/Escherichia_coli_K12.fasta.gz. First start by uncompress the files
```bash
Uncompress annotation file
gunzip ../data/Escherichia_coli_K_12_MG1655.annotation.fixed.gtf.gz
Uncompress genome file
gunzip ../data/Escherichia_coli_K12.fasta.gz
2. Create a file suitable for annotatePeaks.pl.
```bash
cut -f1-5 ../05-PeakCalling/idr/FNR_anaerobic_idr_peaks.bed | \
awk -F "\t" '{print $0"\t+"}' > FNR_anaerobic_idr_peaks.bed
- Try annotatePeaks.pl
```bash
First load bedtools
module add homer/4.10
run Homer annotatePeaks
annotatePeaks.pl –help
Let's see the parameters:
annotatePeaks.pl peak/BEDfile genome > outputfile
User defined annotation files (default is UCSC refGene annotation):
annotatePeaks.pl accepts GTF (gene transfer formatted) files to annotate positions relative
to custom annotations, such as those from de novo transcript discovery or Gencode.
-gtf <gtf format file> (Use -gff and -gff3 if appropriate, but GTF is better)
4. Annotation peaks with nearby genes with Homer
```bash
annotatePeaks.pl \
FNR_anaerobic_idr_peaks.bed \
../data/Escherichia_coli_K12.fasta \
-gtf ../data/Escherichia_coli_K_12_MG1655.annotation.fixed.gtf \
> FNR_anaerobic_idr_annotated_peaks.tsv
-
Open an Rstudio tab in Jupyterhub

-
Add gene symbol annotation using R ```R
set working directory
setwd(“/shared/projects/
read the file with peaks annotated with homer
data are loaded into a data frame
sep=”\t”: this is a tab separated file
h=T: there is a line with headers (ie. column names)
d <- read.table(“FNR_anaerobic_idr_annotated_peaks.tsv”, sep=”\t”, h=T)
Load a 2-columns files which contains in the first column gene IDs
and in the second column gene symbols
data are loaded into a data frame
h=F: there is no header line
gene.symbol <- read.table(“../data/Escherichia_coli_K_12_MG1655.annotation.tsv.gz”, h=F)
Merge the 2 data frames based on a common field
by.x gives the columns name in which the common field is for the d data frame
by.y gives the columns name in which the common field is for the gene.symbol data frame
d contains several columns with no information. We select only interesting columns
d.annot <- merge(d[,c(1,2,3,4,5,6,8,10,11)], gene.symbol, by.x=”Nearest.PromoterID”, by.y=”V1”)
Change column names of the resulting data frame
colnames(d.annot)[2] <- “PeakID” # name the 2d column of the new file “PeakID” colnames(d.annot)[dim(d.annot)[2]] <- “Gene.Symbol”
output the merged data frame to a file named “FNR_anaerobic_idr_final_peaks_annotation.tsv”
col.names=T: output column names
row.names=F: don’t output row names
sep=”\t”: table fields are separated by tabs
quote=F: don’t put quote around text.
write.table(d.annot, “FNR_anaerobic_idr_final_peaks_annotation.tsv”, col.names=T, row.names=F, sep=”\t”, quote=F)
**What information is listed in each column of the file?**
**In which column number is the official gene symbol of the nearest gene?**
**What are all the possible gene types?**
7. Retrieve the list of closest genes
```bash
tail -n 2 FNR_anaerobic_idr_final_peaks_annotation.tsv | awk '{print $11}'
- Count occurences of each associated feature type
# sort | uniq -c to list and count occurences of each item tail -n +2 FNR_anaerobic_idr_final_peaks_annotation.tsv | awk '{print $8}' | sort | uniq -c
How many peaks are associated with promoters ?
tail -n +2 FNR_anaerobic_idr_final_peaks_annotation.tsv | awk '{if ($8=="promoter-TSS") print $11}'
Is the number of genes in your file consistent with the previous reply?
tail -n +2 FNR_anaerobic_idr_final_peaks_annotation.tsv | awk '{if ($8=="promoter-TSS") print $11}' | wc -l
tail -n +2 FNR_anaerobic_idr_final_peaks_annotation.tsv | awk '{if ($8=="promoter-TSS") print $11}' \
> FNR_anaerobic_idr_final_peaks_annotation_officialGeneSymbols.tsv
- Compress back the annotation file
## Compress annotation file gzip ../data/Escherichia_coli_K_12_MG1655.annotation.fixed.gtf
Go back to working home directory (i.e /shared/projects/training/<login>/EBAII2021_chipseq)
## If you are in 07-PeakAnnotation
cd ..
2- Search for Biological Processes, Molecular Functions or Cellular Compartments enrichment
This gene list can then be used with Gene Ontology search tools such as Database for Annotation, Visualization and Integrated Discovery (DAVID) or Ingenuity Pathway Analysis (IPA).
Input your gene list (filename : FNR_anaerobic_idr_final_peaks_annotation_officialGeneSymbols.tsv) on the DAVID website: https://david.ncifcrf.gov/
Are there biological processes enriched in the list of genes associated to the peaks?
Are these genes enriched in some KEGG map?
Bonus: Annotation of ChIP-peaks using R tools
In this part, we will use a different set of peaks obtained using a peak caller from a set of p300 ChIP-seq experiments in different mouse embryonic tissues (midbrain, forebrain and limb).
1 - Obtain the bed files from GEO
From now on, we will work locally on your personal machine.
- We will download the already called peak files in bed format from GEO.
Create a new folder and go in it.
cd /shared/projects/<your_project>/EBAII2021_chipseq mkdir 07-PeakAnnotation-bonus cd 07-PeakAnnotation-bonus - Search for the dataset GSE13845 either using Google or from the front page of GEO
- On the description page, find the three GSM files, and click on each of then
- On each page, select and download the
GSMxxxxx_p300_peaks.txt.gzfile to the newly created folder (wherexxxxxrepresents the GSM number) You should now have downloaded 3 files:GSM348064_p300_peaks.txt.gz (Forebrain) GSM348065_p300_peaks.txt.gz (Midbrain) GSM348066_p300_peaks.txt.gz (limb)
Beware: Make sure to check which genome version was used to call the peaks (remember: this is mouse data!)
2 - Performing a first evaluation of peak sets using R
Now, we will use RStudio to perform the rest of the analysis in R. For the analysis, we will need some R/Bioconductor libraries
- ChIPSeeker
- mouse gene annotation
- mouse functional annotation
- clusterProfiler: Gene set annotation tool
- Go to Rstudio
# load the required libraries library(RColorBrewer) library(ChIPseeker) library(TxDb.Mmusculus.UCSC.mm9.knownGene) library(org.Mm.eg.db) # define the annotation of the mouse genome txdb = TxDb.Mmusculus.UCSC.mm9.knownGene # define colors col = brewer.pal(9,'Set1') - read the peak files for the three datasets:
# set the working directory to the folder in which the peaks are stored
setwd("/shared/projects/<your_project>/EBAII2021_chipseq/07-PeakAnnotation-bonus")
# read the peaks for each dataset
peaks.forebrain = readPeakFile('GSM348064_p300_peaks.txt.gz')
peaks.midbrain = readPeakFile('GSM348065_p300_peaks.txt.gz')
peaks.limb = readPeakFile('GSM348066_p300_peaks.txt.gz')
# create a list containing all the peak sets
all.peaks = list(forebrain=peaks.forebrain,
midbrain=peaks.midbrain,
limb=peaks.limb)
The peaks are stored as GenomicRanges object; this is an R format which look like the bed format, but is optimized in terms of memory requirements and speed of execution.
We can start by computing some basic statistics on the peak sets.
How many peaks?
# check the number of peaks for the forebrain dataset
length(peaks.forebrain)
# compute the number of peaks for all datasets using the list object
sapply(all.peaks,length)
# display this as a barplot
barplot(sapply(all.peaks,length),col=col)
How large are these peaks?
# statistics on the peak length for forebrain
summary(width(peaks.forebrain))
# size distribution of the peaks
peaks.width = lapply(all.peaks,width)
lapply(peaks.width,summary)
# boxplot of the sizes
boxplot(peaks.width,col=col)
What is the score of these peaks?
Can you adapt the previous code to display a boxplot of the peak score distribution for the Forebrain peak set (column Maximum.Peak.Height)?
Where are the peaks located?
We can now display the genomic distribution of the peaks along the chromosomes, including the peak scores, using the covplot function from ChIPSeeker:
# genome wide distribution
covplot(peaks.forebrain, weightCol="Maximum.Peak.Height")
Exercice: use the option “lower” in covplot to display only the peaks with a score (Max.Peak.Height) above 10
How does the signal look like at TSS?
In addition to the genome wide plot, we can check if there is a tendency for the peaks to be located close to gene promoters.
# define gene promoters
promoter = getPromoters(TxDb=txdb, upstream=5000, downstream=5000)
# compute the density of peaks within the promoter regions
tagMatrix = getTagMatrix(peaks.limb, windows=promoter)
# plot the density
tagHeatmap(tagMatrix, xlim=c(-5000, 5000), color="red")
3 - Functional annotation of the peaks
We can now assign the peaks to the closest genes and genomic compartments (introns, exons, promoters, distal regions, etc…)
This is done using the function annotatePeak which compares the peak files with the annotation file of the mouse genome. This function returns
a complex object which contains all this information.
peakAnno.forebrain = annotatePeak(peaks.forebrain, tssRegion=c(-3000, 3000), TxDb=txdb, annoDb="org.Mm.eg.db")
peakAnno.midbrain = annotatePeak(peaks.midbrain, tssRegion=c(-3000, 3000), TxDb=txdb, annoDb="org.Mm.eg.db")
peakAnno.limb = annotatePeak(peaks.limb, tssRegion=c(-3000, 3000), TxDb=txdb, annoDb="org.Mm.eg.db")
genomic localization
We can now analyze more in details the localization of the peaks (introns, exons, promoters, distal regions,…)
# distribution of genomic compartments for forebrain peaks
plotAnnoPie(peakAnno.forebrain)
# for all the peaks
plotAnnoBar(list(forebrain=peakAnno.forebrain, midbrain=peakAnno.midbrain,limb=peakAnno.limb))
Question: do you see differences between the three peak sets?
functional annotation
An important step in ChIP-seq analysis is to interpret genes that are located close to the ChIP peaks. Hence, we need to
- assign genes to peaks
- compute functional enrichments of the target genes.
Beware: By doing so, we assume that the target gene of the peak is always the closest one. Hi-C/4C analysis have shown that in higher eukaryotes, this is not always the case. However, in the absence of data on the real target gene of ChIP-peaks, we can work with this approximation.
We will compute the enrichment of the Gene Ontology “Biological Process” categories in the set of putative target genes.
# load the library
library(clusterProfiler)
# define the list of all mouse genes as a universe for the enrichment analysis
universe = mappedkeys(org.Mm.egACCNUM)
## extract the gene IDs of the forebrain target genes
genes.forebrain = peakAnno.forebrain@anno$geneId
ego.forebrain = enrichGO(gene = genes.forebrain,
universe = universe,
OrgDb = org.Mm.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)
# display the results as barplots
barplot(ego.forebrain,showCategory=10)
Question: do you see an enrichment of the expected categories? What does the x-axis mean? What does the color mean?
Exercise: redo this analysis for the limb dataset and check if the enriched categories make sense.
FAQ
How to extract peaks from the supplementary data of a publication ?
The processed peaks (BED file) is sometimes available on the GEO website, or in supplementary data. Unfortunately, most of the time, the peak coordinates are embedded into supplementary tables and thus not usable “as is”. This is the case for the studied article. To be able to use these peaks (visualize them in a genome browser, compare them with the peaks found with another program, perform downstream analyses…), you will need to (re)-create a BED file from the information available. Here, Table S5 provides the coordinates of the summit of the peaks. The coordinates are for the same assembly as we used.
- copy/paste the first column into a new file, and save it as retained_peaks.txt
- use a PERL command (or awk if you know this language) to create a BED-formatted file. As we need start and end coordinates, we will arbitrarily take +/-50bp around the summit.
perl -lane 'print "gi|49175990|ref|NC_000913.2|\t".($F[0]-50)."\t".($F[0]+50)."\t" ' retained_peaks.txt > retained_peaks.bed - The BED file looks like this:
gi 49175990 ref NC_000913.2 120 220 gi 49175990 ref NC_000913.2 20536 20636 gi 49175990 ref NC_000913.2 29565 29665 gi 49175990 ref NC_000913.2 34015 34115 - Depending on the available information, the command will be different.
How to obtain the annotation (=Gene) GTF file for IGV?
Annotation files can be found on genome websites, NCBI FTP server, Ensembl, … However, IGV required GFF format, or BED format, which are often not directly available. Here, I downloaded the annotation from the UCSC Table browser as “Escherichia_coli_K_12_MG1655.annotation.gtf”. Then, I changed the “chr” to the name of our genome with the following PERL command:
perl -pe 's/^chr/gi\|49175990\|ref\|NC_000913.2\|/' Escherichia_coli_K_12_MG1655.annotation.gtf > Escherichia_coli_K_12_MG1655.annotation.fixed.gtf
This file will work directly in IGV
References
Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., and Gingeras, T.R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. Langmead, B., and Salzberg, S.L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. Langmead, B., Trapnell, C., Pop, M., and Salzberg, S.L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. Ramírez, F., Ryan, D.P., Grüning, B., Bhardwaj, V., Kilpert, F., Richter, A.S., Heyne, S., Dündar, F., and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165.